
Síndrome de Angelman
El síndrome de Angelman (SA) es un trastorno neurogenético poco común que afecta aproximadamente a una de cada 15.000 personas, unas 500.000 personas en todo el mundo. Los síntomas incluyen dificultad para succionar y comer, problemas gastrointestinales, discapacidad intelectual severa, retrasos severos en el desarrollo motor, deterioro del equilibrio y motor y convulsiones.
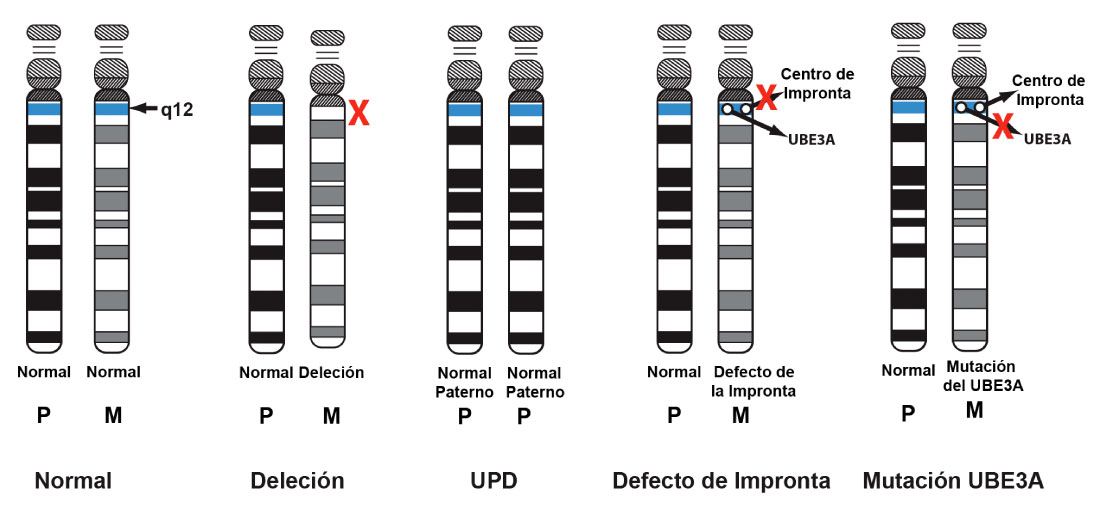
Cada ser humano hereda dos copias de cada cromosoma de sus progenitores, uno del padre y otro de la madre. En principio, se heredan también dos dotaciones de genes; es decir, recibimos dos copias idénticas de cada gen, una del padre y otra de la madre, y ambas copias se expresan y funcionan.
Sin embargo, existen algunos genes que no siguen este patrón. Lo que ocurre es que solamente se expresan en una de las copias, la paterna o la materna; en lenguaje coloquial, una de las copias funciona y la otra no (está inactiva o silenciada). Estos genes un tanto especiales se dice que están sometidos a impronta genómica.
En el Síndrome de Angelman el cromosoma que se ve afectado es el número 15. En ese cromosoma se encuentra un gen llamado UBE3A (se encuentra en una región concreta del cromosoma, en la 15q11-q13). Pues bien, este gen es uno de los que presenta impronta genómica. El mecanismo por el que se produce el SA es el siguiente:
- Una persona sana recibirá dos copias del cromosoma 15 y dentro de él dos copias del gen UBE3A: la copia de este gen heredada del padre estará inactiva o silenciada, mientras que la que está activa y funciona es la copia heredada de la madre.
En una persona afectada por el SA, el gen UBE3A heredado de la madre no se expresa, no funciona correctamente. Y no puede compensarse con la copia paterna, porque ésta está inactiva siempre para este gen.
